

DNA储存着我们的遗传信息,然而在细胞中真正执行功能的是蛋白质。每个蛋白质的氨基酸链扭曲、折叠、缠绕成复杂的结构,“看清”它们的结构对理解其功能至关重要。但想要破解这种结构通常需要花很长的时间,有些甚至难以完成。
“用机器学习去研究蛋白质结构预测,在这个领域属于少数派。一直到2016年,甚至到2018年,这个领域大部分人都还在试图用能量优化,而不是机器学习或者深度学习去研究这个问题。”美国芝加哥丰田计算技术研究所教授、北京大学客座教授许锦波在接受澎湃新闻(www.thepaper.cn)记者专访时如是表示。
许锦波被业界誉为“AI预测蛋白质结构全球第一人”。早在2016年,他开发的RaptorX-Contact方法,首次证明了深度学习方法预测蛋白质结构的可行性,让始终在“门口”徘徊的蛋白质结构预测终于迈出实质性的一步,也自此掀起了AI蛋白质结构预测的热潮。

美国芝加哥丰田计算技术研究所教授、北京大学客座教授许锦波。
现年48岁的许锦波从小就是一名不折不扣的“学霸”。1990,16岁的许锦波在全国高中数学联赛中获江西赛区第一名,这也是当时江西临川县首次获得该类奖项殊荣。1991年,因为在数学竞赛中的优异成绩,他从临川一中被保送至中国科学技术大学计算机系,1999年获得中国科学院计算所硕士学位。2003年,许锦波获加拿大滑铁卢大学博士学位,之后任该校研究助理教授、麻省理工学院博士后研究员。
2001年,尚在攻读博士学位的许锦波开始接触计算生物学,彼时的导师提议,“有一个很难的问题,就是研究蛋白质折叠,想不想做?”在此后的二十余年时间里,许锦波研究的重要课题之一就是开发和优化软件,去无限缩小蛋白质结构预测结果和真实构型之间的差距。
近日,在未来论坛主办的2022《理解未来》科学讲座01期“AI+蛋白质结构和功能预测”上,许锦波也首先谈到,其实蛋白质结构预测这个问题已经研究了几十年,过去这个领域一直比较冷清,特别是在2006年到2016年这10年间,“当时大家都觉得这个问题是没办法做出来的,所以很多人都离开这个领域去做其他的问题了。”
这样的冷清已经是过去式。在最近的几年时间里,这一领域陆续获得突破性的进展。2020 年,人工智能预测蛋白质结构也被国际顶级学术期刊《科学》杂志评为十大科学突破之一。“现在人工智能预测蛋白质结构受到的关注,远远超过了过去几十年来的关注。”许锦波表示。
然而,在冷清的路上走惯了的许锦波,对眼下的热闹并没有表现出太多的兴奋。谈及这两年陆续成立的人工智能应用于生命科学领域的公司,他坦言,“我对产业的了解不是很多,也就最近几个月开始接触一些产业界的认识和做投资的人。”当然,许锦波认为,对于“AI For science”的产业化而言,当下的确处于一个比较好的时候。
但许锦波强调,就人工智能预测蛋白质结构而言,重复实现明星公司DeepMind的AlphaFold2不应该成为其他团队的目标,“这种改进只是一个渐进式的改进,并不是一个非常大的突破,这个领域仍然有一系列问题真正需要我们去解决。”对于人工智能在药物研发等生命领域的应用,他则表示,“希望能够做出一些真正有用的东西出来。”
始于半个世纪前的猜测
蛋白质结构预测,始于科学家们的一种设想,是否无需实验就能获取蛋白质的三维结构?
在蛋白质结构解析的几十年历史中,结构生物学家们用X射线晶体学、核磁共振波谱学(NMR)、冷冻电镜(Cryo-SEM)技术解析了很多蛋白的结构,并以此更好地推进疾病机理、药物研发等工作。
然而,这些手段被视作劳心劳力又价格高昂。截至目前,约有10万个蛋白质的结构已经用实验方法得到了解析,但这在已经测序的数10亿计的蛋白质中只占了很小一部分。
作为学计算机出身的一名科学家,许锦波对他研究了近20年的蛋白质如此理解:蛋白质是由很多氨基酸通过化学键串联在一起,如果把每个氨基酸看成一个珠子的话,那么就有20种不同颜色的珠子,这些珠子串在一起形成蛋白质的氨基酸系列,每一个不同的颜色用一个字母表示,所以蛋白质氨基酸序列可以看成是1个由20个字母组成的字符串。每个氨基酸又是由几十个原子组成的,所以整个蛋白质是由成千上万个原子构成的,这些原子在细胞里面有相互作用力,最后形成一个稳定的构型。
“我们可以用不同的软件去把这些结构给显示出来,但是在利用这些软件去显示蛋白质构型的时候,我们需要知道这些原子在三维空间中的位置,需要知道它们的三维坐标,怎么样才能知道这些三维坐标?”许锦波提到,在过去很多年里,科学家发展了三种主要的实验技术,去测定这些原子的三维坐标。
除了上述提到的三种实验室技术之外,科学家们也在研究,计算方法的推导是否可行?
许锦波对澎湃新闻记者表示,美国生物化学家、1972年诺贝尔化学奖得主克里斯蒂安·安芬森(Christian Boehmer Anfinsen)通过实验提出了他自己的猜想,“这位实验学家的猜测基本是对的,他自己做了一些列实验支持了这个理论。”
安芬森的工作大部分围绕蛋白质的结构与功能之间的关联性。1961年,他研究了核糖核酸酶可以在变性后重新进行折叠,恢复到原来的空间结构,同时保留酵素的活性。安芬森因此认为,所有造成最终构象所需的蛋白质信息都被编码于其氨基酸序列上,即蛋白质一级排序决定三维结构。
上述即被称为安芬森法则,这也是蛋白质结构预测的基石。
美国生物化学家、1972年诺贝尔化学奖得主克里斯蒂安·安芬森。
然而,在随后的50多年时间里,科学家们使用了各种各种的方法,都无法精确计算蛋白质的三维结构。“在安芬森这个假设和理论基础之下,科学家们去做蛋白质折叠预测,都是从能量优化的角度去做。”许锦波解释,大家普遍认为,蛋白质是折叠到最小能量状态,这也意味着,从理论上来说,如果能更好地优化这个能量函数,就能够找到蛋白质的最小能量状态。
但这一思路有着天然缺陷。“第一,一个蛋白质是一个非常大的体系,由成千上万个原子组成,对应一个非常巨大的搜索空间,构型是千变万化的。”许锦波继续提出第二个困难之处,“虽然说大家普遍接受蛋白质折叠到最小能量状态,但能量函数到底是什么样的?我们本身就对能量函数的理解还不是特别好。”
许锦波在博士阶段最初也是使用传统的优化算法去研究这一问题。2001年,他接下了导师向他抛出的这一课题,第二年即取得了不错的成果,在2002年全球蛋白质结构预测比赛CAFASP(用于全自动高通量蛋白质结构预测的评比)中,夺得冠军。
回忆当时的成绩,许锦波略显轻描淡写,“虽然排名最好,但是意义并没有那么大,并没有改变这个问题的现状,只是结果比别人好一点点而已。”在这一思路下继续了一年多之后,他意识到,传统的优化算法可能不是一个很好的路径。
2006年,许锦波开始转向机器学习,彼时已组建独立实验室的他认为,应该改变策略。“我们用机器学习做的比传统的方法好一点,在蛋白质结构预测比赛中,也取得了很好的成绩,比别的组要好一点,但也并没有特别大的改变。”
这条路径一走就又是8年,应该也是许锦波科研道路上最冷清的8年,很多人陆续转行,领域几无关注。
人工智能为什么可以成功
2014年,许锦波开始第二次转换途径。
“2012年,深度学习开始在图像识别中做到了很好的结果,所以我们在2014年开始尝试用深度学习去研究这个问题。”真正将AI纳入到许锦波预测蛋白质结构的工具箱中,始于这一年。彼时,同领域中只有极少数人关注到这一新的工具。
“新方法不是去做能量最优化,而是预测原子之间的相互作用关系。”
许锦波进一步解释道,假设已有一个氨基酸序列,那么把和这一蛋白质同源(同一个家族)的那些蛋白质都找出来,然后把所有这些同一个家族的蛋白质的氨基酸序列都比对在一起。“多序列对比下,我们用矩阵去表示蛋白质里面氨基酸之间相互作用关系,然后根据相互作用关系矩阵,就可以把蛋白质的原子的坐标预测出来,这是这种新方法的总体思路。”
当然,在总体思路框架下可以有不同的实现方法,“但新方法的关键点在于,我们能不能准确地推断出蛋白质里面原子之间或者氨基酸之间的相互作用关系,这一步是非常关键的。”
许锦波谈到,为了预测原子之间的相互作用关系,科学家们探索的最早方法是协同进化全局统计方法(global statistical method for co-evolution analysis)。然而,这种方法只对极少比例蛋白质有效,而往往这些蛋白质家族里某些蛋白的三维结构已经被实验技术测出来了,这也意味着用这种方法预测的意义并不太大。
他认为,真正对大量的蛋白质结构预测其作用的转折之年是2016年。在转向深度学习2年之际,许锦波开始用深度学习预测蛋白质的三维结构。而在此前的2年时间里,其团队以更为简单的问题入手,尝试预测蛋白质的二级结构,即肽链主链骨架原子的空间位置排布,不涉及氨基酸残基侧链。
“对于这么一个简单的问题能够做得好,我们认为对于更难的问题,也就是预测蛋白质的三维结构应该会有效果。”许锦波提到一个细节,在2015年其就组织学生去解决三维结构的问题,然而并没有实现,“他们不太理解我的想法,因为那个时候在这个领域没有人用深度卷积网络去解决这个问题。”
2016年,腾出一些时间的许锦波开始自己写代码去实现自己的算法,“大概在那年暑假的时候就得到了非常好的结果,发现一下子能做得比以前的方法好非常多,2016年秋天,我把结果写成一篇论文发布在了网上。”发布后的第一个月,即在领域内引起了一波关注高潮。
许锦波发布的正是他开发的第一代人工智能方法RaptorX。该方法基本的原理是,通过深度卷积残差网络(ResNet),对蛋白质的序列进行卷积变换,从中抽取出有效信息,同时也对蛋白质残基之间相互作用关系进行卷积变换。通过这两者不同的卷积变换,可以非常准确地预测蛋白质氨基酸之间的相互作用关系。“然后基于这个相互作用关系,我们可以把它的三维结构重构出来。”
在2016年全球蛋白质结构预测比赛(CASP12)中,这一尚未完善好的方法即崭露头角,“当时已经做得非常好,做的比其他传统方法都要好。”

2017年1月,许锦波将前期成果正式发表于《PLOS Computational Biology 》,题为“Accurate De Novo Prediction of Protein Contact Map by Ultra-Deep Learning Model”。在这篇论文中,研究团队展示了通过使用深度残差卷积网络,可以大幅度提高蛋白质预测的精度,并且这种学习方法也很容易推广到不同类型的蛋白质层面,比如一些膜蛋白及蛋白复合物等的结构。
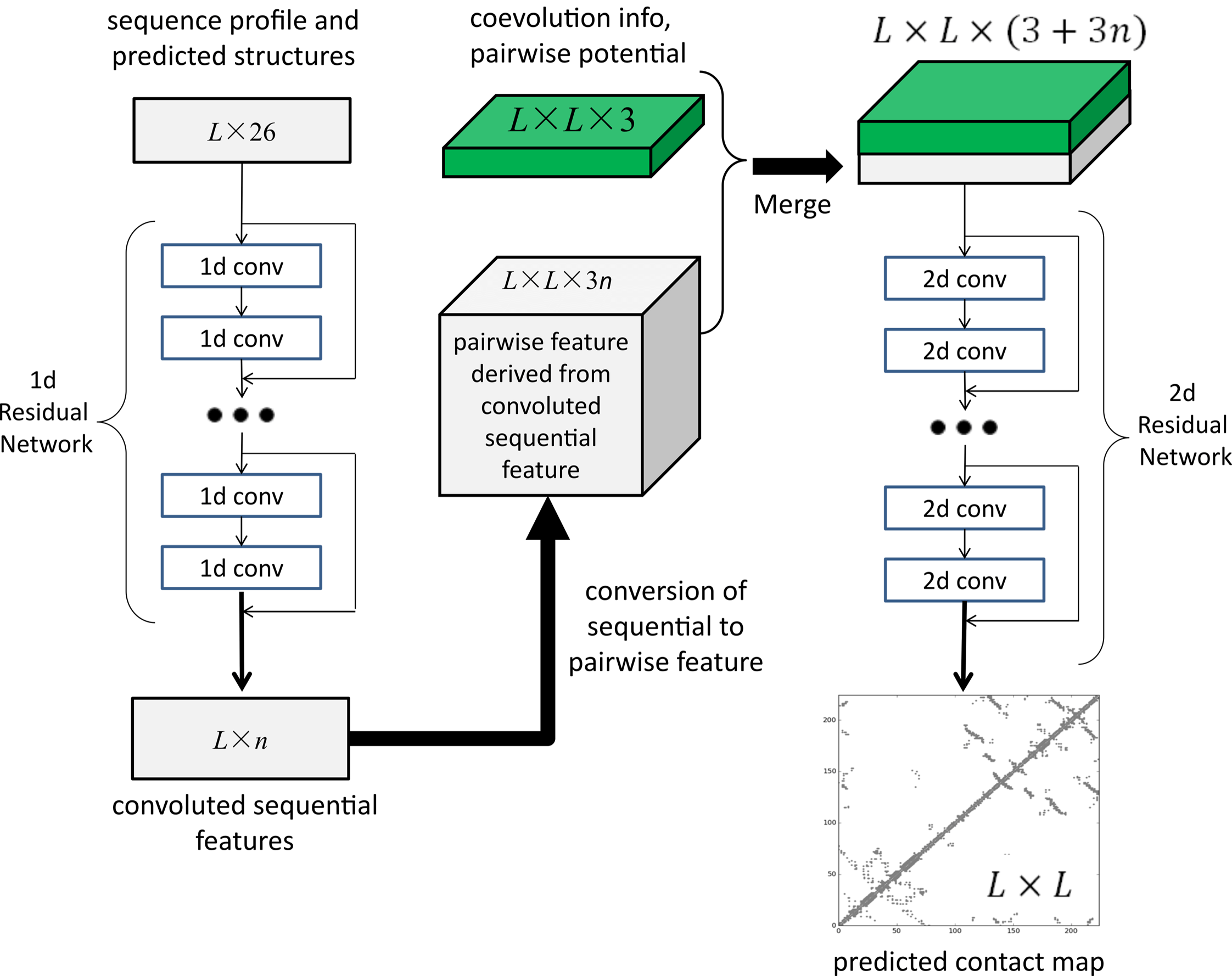
至今这仍是许锦波最满意的一篇论文。“我们论文出来之后,其实把问题定义得很清楚了。从AI的角度来说,就是告诉大家这个问题的输入是什么,输出是什么,你只要把AI算法做好就行了。至于你用什么AI算法,无非更多的是工程上和计算资源上的问题。”
他还向澎湃新闻记者回忆了一段小插曲,研究团队实际上最开始将论文投到了《自然》(Nature)的一本子刊,然而编辑并不太相信他们的结果。“因为这个问题研究很多年了,一直没有什么进展,他不认为我们能做得这么好,另外一本期刊的一个评委都不认为我们的结果是可靠的。”
令许锦波欣慰的是,无论是学术界还是产业界,都在论文发表之后对该研究给予了广泛的关注。他感受到,总体而言,学计算机出身的人更容易接受他们的结果,而学生物化学或者生物物理的人,因为此前就不习惯于使用类似的方法,并不太容易接受这项结果。
值得一提的是,在蛋白质结构预测领域过去近30年的时间里,该领域的发展大致可以分三个阶段。第一个阶段,也就是长达20多年的时间里,在传统方法之下该领域进展非常缓慢;第二个阶段,也就是通过使用许锦波等人开发的第一代人工智能方法RaptorX,难度较大的蛋白质结构的预测精度已被大幅提升;而在第三个阶段,则是目前为止全球表现最好的蛋白质结构预测工具,也就是DeepMind在2020年推出的AlphaFold2。“通过使用注意力机制网络,又可以大幅度提高蛋白质结构预测的精度。”
在许锦波看来,DeepMind在2017年、2018年之际,实际上在重新实现他的算法,“当然他们工程上做得比我们好一些。”而对于DeepMind在AlphaFold2中使用的注意力机制网络,其最早被应用于自然语言处理中。
“计算生物学领域的人知道的并不是很多,最早将这一网络真正用到这个领域的是Facebook,他们没有用来做蛋白质结构预测,而是用来对蛋白质序列进行建模。”许锦波提到,即使后来计算生物学领域的人注意到了基于注意力机制的网络,然而该网络需要太多的计算资源,“学术界没有人有这么多资源去做这件事情。”
许锦波坦言,其团队在2020年曾经考虑如何简化基于注意力机制的网络,“希望使它能够在我们的计算资源上跑起来,这是我当时做的事情,因为我们没有几百块GPU(显卡上的芯片)。”相比之下,背靠谷歌的DeepMind完全没有这方面的“资源窘境”,可以用很多GPU卡训练他们的模型。
许锦波认为,从思想创新而言,AlphaFold2迈的这一步并不没有让人感到非常吃惊的。“真正吃惊的是他们能够一下子调动30个人去做这个事情,能够把它实现得非常好,我觉得这是他们的长处。”
总体而言,人工智能对蛋白质结构预测领域起到了非常大的推动作用,而过去这么多年里,为何又只有深度学习能够做到?
许锦波分享了他个人的理解,首要的前提是,深度学习是基于现有的理论基础,特别是进化论。“第一,虽然我们没有它们的构型,但是我们知道,同一个家族的蛋白质结构应该是很相似的。第二,同一个蛋白里面空间中相邻的氨基酸互相影响、共同进化,这点也非常重要。”
除理论基础外,许锦波认为对于训练深度学习算法而言,数据当然必不可少。“现在我们有了大量的蛋白质序列数据,可以依据同一个家族里面蛋白质的进化关系去推断原子在空间中的距离,这是非常重要的。另外一个很重要的数据源是我们也有了一些蛋白质结构数据,虽然说没有那么多,但现在我们至少有一些,那么通过指导深度学习模型去学习氨基酸共进化与原子间中距离的关系。”
比重复实现AlphaFold2更重要的事情
尤其在AlphaFold2出现之后,人工智能预测蛋白质结构这一领域受到了空前的关注,终于“热闹”了起来。
许锦波总结认为,人工智能的确颠覆了蛋白质结构预测,而这会带来非常大的改变,尤其对分子生物学科来说,“我想这个结果现在已经改变了很多分子生物学家的研究范式,以前的分子生物学家基本都基于蛋白质的氨基酸序列去分析蛋白质的功能,现在很多人都开始使用预测的结构去做研究、去分析蛋白质的功能,所以这是一个非常大的研究范式的改变。”
但现在还远远没有到达终点,将来又如何继续推进人工智能在结构生物学甚至更广泛的生物学中的应用?
许锦波谈道,有很多团队在致力于重复实现AlphaFold2,“当然这是一条必经之路,但这种改进只是一种渐进式的改进,即使我们能够做的好一点点,其实也不是一个非常大的突破。”他同时提醒,如果很多团队或者初创公司一窝蜂去做这件事情,“我觉得有点浪费资源。”
在他看来,那些当下解决得还不够好的问题,需要去真正地投入更多的精力。
例如,我们能不能对一个孤儿蛋白进行非常准确预测?能不能预测蛋白质的折叠过程,而不仅仅是最后构型?能不能准确预测蛋白质复合物或者一个多域蛋白的结构?能不能预测蛋白质和多肽、DNA或者RNA的相互作用?能不能预测单点或多点突变对一个蛋白质结构和功能的影响?
他对澎湃新闻记者进一步表示,我们对蛋白质结构预测的要求取决于我们的目标。如果目标只是想知道这个蛋白质最终的三维形状,对于大部分蛋白质来说其实已经做到了这一点。“然而现在我们能做的,就是可以把单个蛋白的结构预测得很好。但是对于蛋白质复合物等更加复杂的情况,人工智能的方法确实能做得比以前好很多,但是还没有达到让人非常满意的状态,这个方向还需要花更多的时间去研究。”
许锦波同时抛出一个更值得思考的问题,“现在所有的成功方法其实都有点cheating。”这也是一个从原理上即存在的问题。
不难理解,如此说的原因在于,目前的方法需要使用大量的蛋白质同源信息,“能够找到越多的同源蛋白,这种预测效果越好。如果没有这部分的信息,现在所有的方法都没有效果。”许锦波说,在细胞里面,也就是自然界的蛋白质在折叠的时候,“它并不知道同家族到底有哪些蛋白质,它自己能够折叠出来,它不需要知道有多少‘兄弟姐妹’。”
值得一提的是,许锦波已经回国,并决定将重心转移到国内。“创新驱动发展战略是我们国家综合国力发展的有力保障,”许锦波对澎湃新闻记者表示,“我希望做一些真正原创且能落地的东西出来,推动科研与产业化的融合发展。”
谈到“AI+生命科学”的产业应用价值,许锦波表示,目前“AI for Science”的产业化环境很好,特别是“AI for BioTech”。“国家在‘AI for BioTech’领域非常重视,投资机构也非常支持硬科技领域的早期、长期投资。”而从产业角度来讲,他认为,由于AI在生物制药领域为各个环节赋能,帮助行业提升了效率与准确度,因此AI在该领域的产业化也具有很好的前景。
值得关注的是,今年1月,许锦波在北京创立北京分子之心科技有限公司(下称“分子之心”)。就在4月,该公司宣布已完成数千万美元天使轮融资,由红杉中国领投,百度风投、生命园创投基金、NeuX Capital芯航资本 、未来启创基金等跟投。分子之心称,该轮融资将用于进一步扩大团队、AI蛋白质平台的持续进化,以及科研成果的产品化转化。
他对澎湃新闻记者表示,公司目前仅有一个很小的团队在继续研究蛋白质结构预测的问题,“我们更主要的目标在于,能不能做各种蛋白质的优化和设计。比如可以把一个抗体优化得更好,使得它能够跟抗原结合更好;或者说能不能设计一个自然界不存在的蛋白,用它来做药或用于其他目的;或者能不能把某一个酶优化得更好。这是现在我们公司的重点。”
其最后谈到,当下多学科的融合比以往更加重要,而如何吸引更多的人加入到交叉学科,同时也吸引更多的学生进入到领域内,这些仍面临一些挑战。
许锦波以其自身经历说道,“刚进入计算生物学这个领域的时候,我会发现我跟生物学家们的沟通其实是非常困难的。只有经过一段时间之后,谈话和合作才能继续下去,多沟通多交流,我想这是非常重要的。”
而更为关键的一点是,他认为评估体系应当做出一些改变。“从我的经历来看,做蛋白质结构预测或者说做计算生物学,以前其实不太受重视。之前论文都发表不到特别高影响因子的刊物上,而影响因子又跟这个领域多少人在做有关系。如果你用影响因子去评估一项计算生物学的工作的话,往往这些人是比较吃亏的,也进而打压了那些做计算生物学的学生。”
许锦波的观点是,大家应当以比较开放的心态,容忍不同领域人的发展。
·本文经授权转载自澎湃新闻
·原文链接:https://www.thepaper.cn/newsDetail_forward_17917804




